Database searching with MS/MS
1. For shot-gun sequencing method, what input information do you need for a successful database search?
fragmentation spectrum
- precursor or peptide mass
- peptide charge state
search parameters
2. Can you list possible steps/methods for database reduction in the precursor approach?
Precursor mass filter approach
- Database is initially filtered to only include peptide sequences matching the parent ion m/z
- Empirical and theoretical fragment ions are compared for all parents satisfying the parent m/z and best scoring matches are reported
Sequence tag approach
- Short stretches of possible amino acid sequence are deduced by de novo sequencing approach
- All possible peptides that could contain those sequences are selected from database to be compared and scored
- Does not consider parent ion m/z in search
3. On slide No.14, please rationalize different rankings (good to terrible) of these MS/MS spectra.
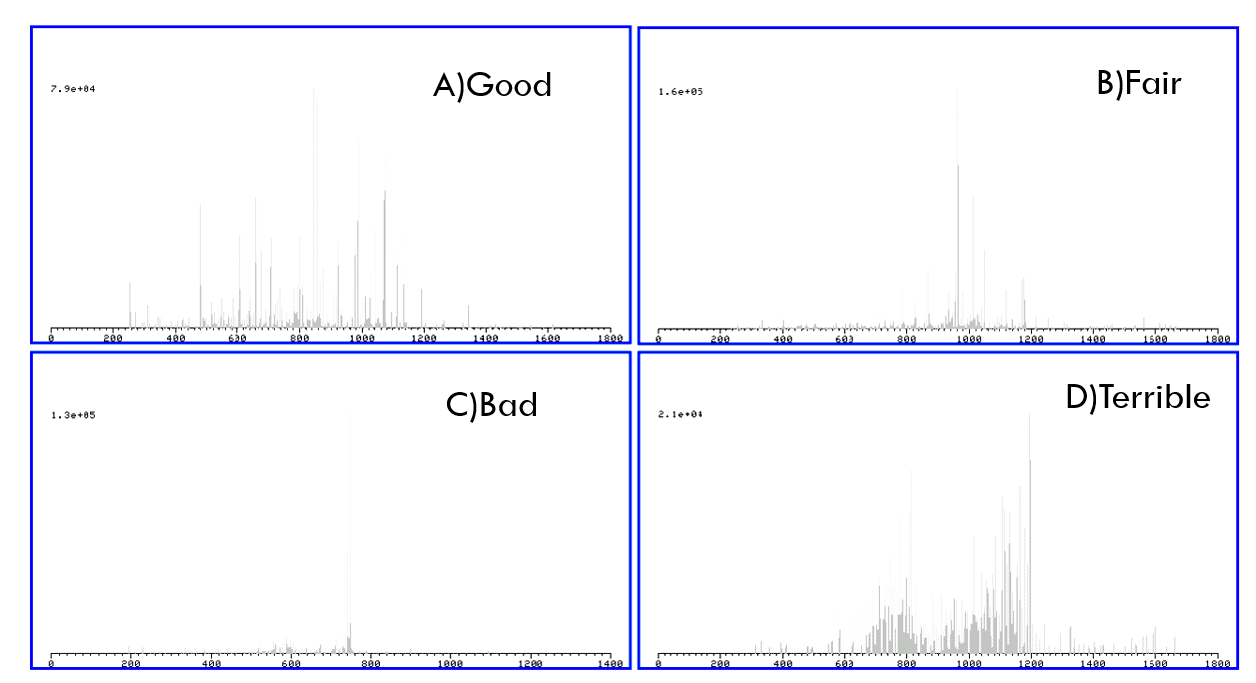
Good: High mass resolution & high signals & low noise
Fair: Qualified mass resolution & Weaker signals than good
Bad: Low signals for all
Terrible: Low mass resolution & numerous peaks & loud noise, DISASTER FOR DATABASE SEARCH
4. Decoy database is typically generated by reversing protein dataset. For one search result, if the final identification # is 500 peptides with 1% FDR. It means how many decoy peptides in the results?
2.5
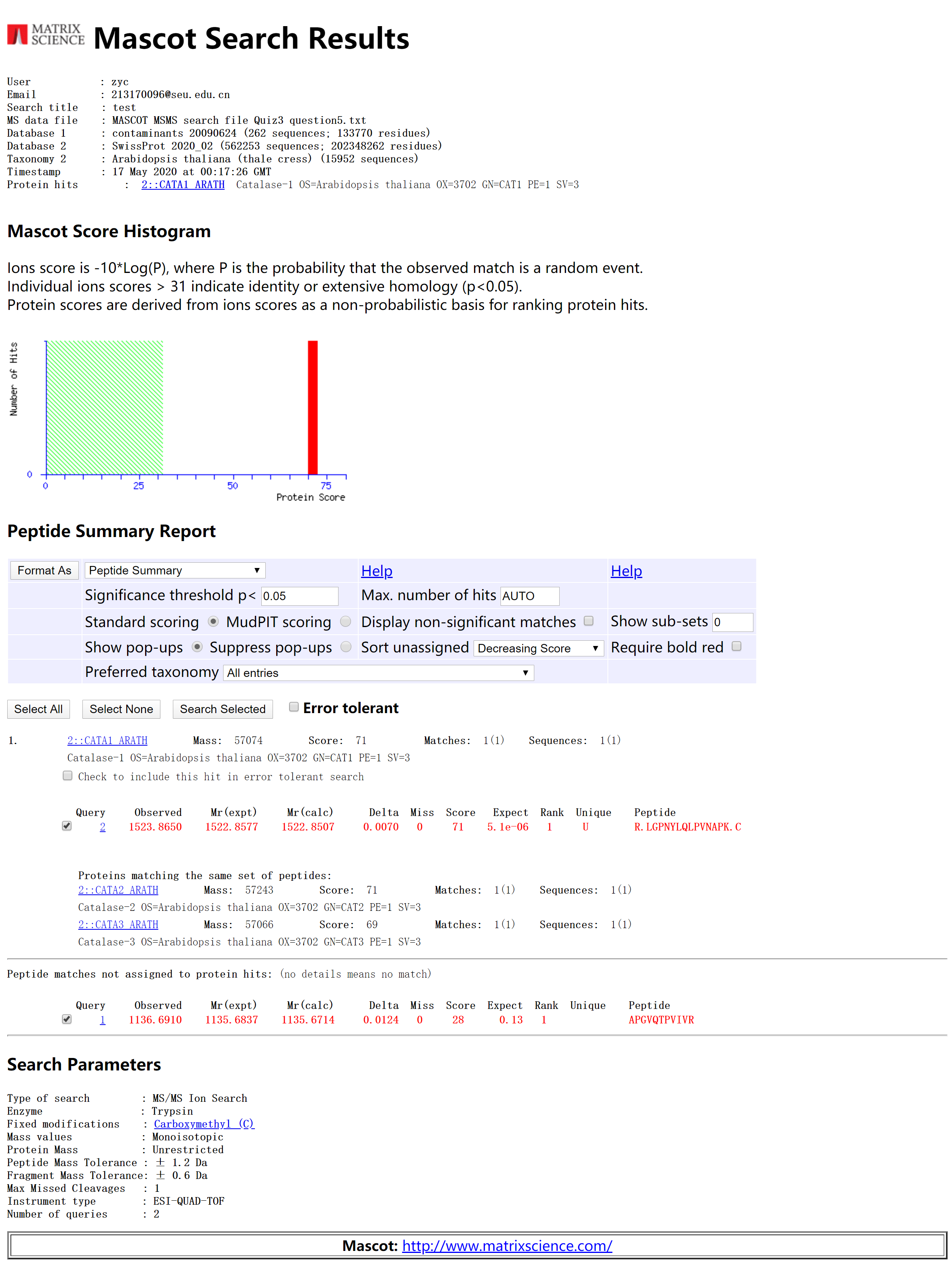
5. The following two tandem MS spectra were obtained on an ESI Q-TOF instrument. These tryptic peptides are from an Arabidopsis thaliana protein. The sample was reduced and alkylated with iodoacetamide prior to digestion (so cysteines are carbamidomethylated). Identify the protein using Mascot. Remember, you will need to use the correct file format (To make it easy, txt file has already generated for you). Make sure you search the data from both spectra simultaneously, and don’t forget about the precursor charge state issue in the data file! Both spectra are from doubly charged parent ions of m/z = 568.8455 (upper spectrum) and m/z = 762.4326 (lower spectrum). Please attach a copy of the first page of your database search results, including search parameters.
PTM analysis by MS
1. Can you use NIH PubMed website to identify one literature each (after 2010) on protein PTM related to protein location, activity state and turn over, respectively?
[1] Shah, R. N., Grzybowski, A. T., Cornett, E. M., Johnstone, A. L., Dickson, B. M., Boone, B. A., Cheek, M. A., Cowles, M. W., Maryanski, D., Meiners, M. J., Tiedemann, R. L., Vaughan, R. M., Arora, N., Sun, Z.-W., Rothbart, S. B., Keogh, M.-C., & Ruthenburg, A. J. (2018). Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Molecular Cell, 72(1), 162–177. https://doi.org/10.1016/j.molcel.2018.08.015
[2] Zheng, N., & Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annual review of biochemistry, 86, 129-157.
[3] Vu, L. D., Gevaert, K., & De Smet, I. (2018). Protein Language: Post-Translational Modifications Talking to Each Other. Trends in Plant Science, 23(12), 1068–1080. https://doi.org/10.1016/j.tplants.2018.09.004
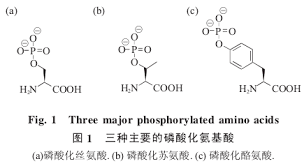
2. What are three amino acids that can be phosphorylated? Please draw the structures of phosphorylated amino acids.
3. Can you list at least FOUR reasons why PTM identification is harder than general protein identification?
I) Sequence coverage becomes an issue
- Need only a fraction of the total sequence to ID a protein
- Often need near 100% coverage to identify all modifications
II) Sometimes specific peptide or sequence needs to be detected
- Don’t care which peptides are detected for protein ID
- Remember, many peptides ionize poorly or are too big or too small
III) Modifications are often dynamic (reversible) and have low stoichiometry (small % of total protein is actually modified at a given site)
- Therefore, signal may be weak for modified peptides
- More sample may be required
IV) Some modifications can decrease ionization of peptides
- Charge (phosphorylation)
- Mass (glycoslyation)
V) Need good fragment ion coverage in many cases to unambiguously identify sites of PTM
4. List at least three methods to enrich phosphopeptides.
Enrichment of subclasses of phosphoproteins:SH2 domains & 14-3-3 domains & Phosphatase trapping mutants
Immunoprecipitation using anti-phosphotyrosine (pY) antibodies
Enrichment of phosphopeptides using IMAC, TiO2, and polymac methods
5. The top MSMS spectrum below is from a parent peptide of m/z = 1266.6 that matches the mass of a predicted peptide from the protein of interest – KLSFHVYEDE. The lower MSMS spectrum is from a parent peptide 80 Da greater, 1346.6, suggesting it could be a phosphorylated form of the top peptide. The data were acquired on a MALDI TOF/TOF instrument. Identify which amino acid in this peptide is phosphorylated and describe which ions allow you to conclusively make that assignment (HINT – use an online software such as Protein Prospector to obtain the predicted fragment ions for this peptide sequence).
The Ser
the b6 peak 693 shifting 80 from 613